علل ژنتیکی کمخونی فانکونی
مهمترین ژن دخیل در کمخونی فانکونی، ژن FANCB است؛ البته در مجموع 14 ژن در بروز این بیماری نقش دارند.- اختلال در ترمیم DNA: جهش در ژنهای فانکونی موجب ناتوانی بدن در ترمیم آسیبهای ژنتیکی میشود.
- تجمع آسیبهای ژنتیکی: سلولهایی که آسیبهای ژنتیکی را به خوبی ترمیم نمیکنند، با گذشت زمان از بین رفته یا عملکردشان مختل میشود؛ این روند در بافتهایی مانند مغز استخوان مشکلات جدی ایجاد میکند.
نقش عوامل محیطی در تشدید بیماری
افراد مبتلا به کمخونی فانکونی حساسیت بالایی نسبت به مواد سمی DNA و برخی اشعهها دارند:- سیسپلاتین و میتومایسین C: داروهای شیمیدرمانی که آسیب DNA را در این بیماران تشدید میکنند.
- UVA (اشعه ماوراءبنفش): مواجهه طولانیمدت با اشعه فرابنفش میتواند خطر ابتلا به سرطان را در این افراد افزایش دهد.
علائم و ویژگیهای بالینی کمخونی فانکونی
اختلالات ظاهری و رشدی
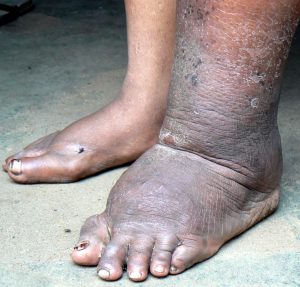
- ویژگیهای چهره: برخی بیماران صورت مثلثیشکل دارند.
- پوست: لکههای تیره یا قهوهای (هیپرپیگمانتاسیون) روی پوست مشاهده میشود.
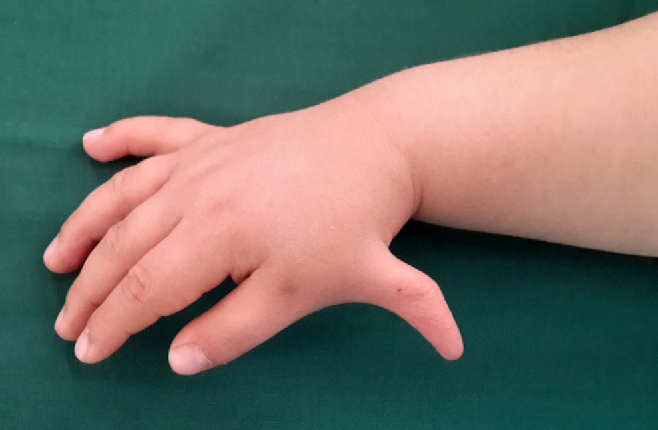
- ناهنجاریهای اسکلتی: کاهش وزن هنگام تولد، کوتاهی قد، استخوانهای غیرطبیعی یا داشتن انگشت اضافه.
- اختلالات رشد: کندی رشد جسمی نسبت به همسنوسالان.
سرطانها و بیماریهای مرتبط
- سرطانهای خون: لوسمی میلوئید حاد (AML) شایعترین است.
- تومورهای جامد: کارسینوم سلولهای سنگفرشی سر و گردن (HNSCC) و سرطانهای دستگاه گوارش، کبد و تخمدان که معمولاً پیش از 50 سالگی بروز میکنند.
مشکلات خونی
- نقص در مغز استخوان: کاهش تولید گلبولهای قرمز و سفید که باعث کمخونی و ضعف سیستم ایمنی میشود.
- خونریزی و عفونت: بهدلیل عدم تولید کافی پلاکتها و سلولهای ایمنی.
- کمخونی آپلاستیک و دیسپلازی مغز استخوان: خطر ابتلا به این اختلالات خونی در افراد مبتلا بیشتر است.
روشهای تشخیص کمخونی فانکونی
- آزمایش شمارش کامل خون (CBC): برای شناسایی کمخونی و بررسی میزان گلبولهای قرمز و سفید.
- آزمایش کروموزومی: بررسی شکستهای کروموزومی با استفاده از داروهای حساسیتزا مانند میتومایسین C (MMC) و دیاتیلبوتان (DEB).
- آزمایش مغز استخوان: ارزیابی میزان تولید سلولهای خونی و بررسی احتمال بروز سرطان در مغز استخوان.
- آزمایشهای ژنتیکی: تشخیص جهشهای ژنی در بیماران و اعضای خانواده برای پیشگیری و مراقبت زودهنگام.
درمان و کنترل کمخونی فانکونی
مشاوره ژنتیکی
- تشخیص ناقلان ژن از طریق آزمایش ژنتیکی اعضای خانواده.
- بررسی پیش از تولد در صورت سابقه خانوادگی کمخونی فانکونی.
روشهای درمانی رایج
- انتقال خون: برای تأمین موقتی سلولهای خونی و کنترل علائم کمخونی.
- داروهای تحریککننده مغز استخوان: داروهایی که تولید گلبولهای قرمز و سفید را افزایش میدهند.
- پیوند مغز استخوان: تنها روش درمانی قطعی در بیماران با نقص شدید مغز استخوان؛ البته امکان افزایش خطر ابتلا به سرطانهای ثانویه وجود دارد.
- درمانهای نوین: پژوهشها در زمینه ویرایش ژن و داروهای ترمیمکننده DNA همچنان ادامه دارد.
عوارض درمان
- افزایش خطر تومورهای جامد پس از پیوند مغز استخوان.
- حساسیت شدید به شیمیدرمانی به علت نقص در سیستم ترمیم DNA.
پیشآگهی و امید به زندگی
بسیاری از بیماران در سنین نوجوانی یا اوایل جوانی (14 تا 25 سالگی) تشخیص داده میشوند. موفقیت پیوند مغز استخوان، بررسیهای منظم جهت تشخیص زودهنگام سرطانها و رعایت مراقبتهای پزشکی همهجانبه، از مهمترین عوامل مؤثر در افزایش طول عمر بیماران هستند. با وجود پیشرفت در روشهای درمانی، مراقبت مادامالعمر و پیگیری منظم در کنترل عوارض و ارتقای کیفیت زندگی این بیماران نقشی حیاتی دارد.پیشگیری از کمخونی فانکونی
- آزمایشهای ژنتیکی پیش از بارداری: برای والدینی که سابقه کمخونی فانکونی در خانواده دارند.
- تشخیص پیش از تولد: بررسی DNA جنین بین هفتههای 10 تا 15 بارداری.
- اجتناب از مواجهه با عوامل سرطانزا: از جمله اشعه ماوراءبنفش و مواد شیمیایی خطرناک، که میتواند بروز سرطان را افزایش دهد.